Hypertrophic Cardiomyopathy (HCM)
HCM Personal Stories




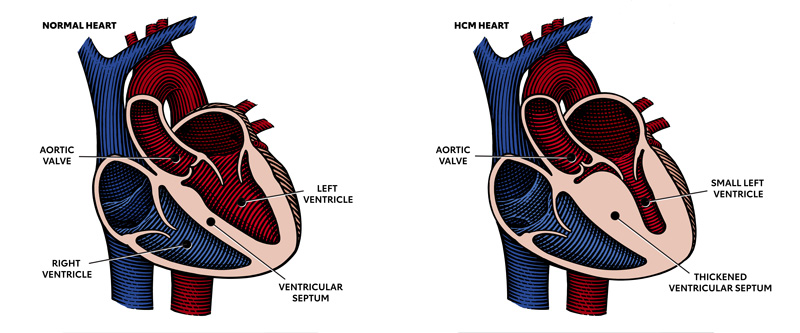
What is hypertrophic cardiomyopathy?
Hypertrophic cardiomyopathy is most often caused by abnormal genes in the heart muscle. These genes cause the walls of the heart chamber (left ventricle) to become thicker than normal.
The thickened walls may become stiff and this can reduce the amount of blood taken in and pumped out to the body with each heartbeat.
View our printable information sheets:

Watch this episode of House Calls: Explaining HCM (Video).
Watch this video for a quick overview of HCM.
Obstructive and nonobstructive HCM
In obstructive HCM, the thickened part of the heart muscle, usually the wall (septum) between the two bottom chambers (ventricles), blocks or reduces the blood flow from the left ventricle to the aorta. Two-thirds of the people with HCM have this type.
In nonobstructive HCM, the heart muscle is thickened but doesn’t block blood flow out of the heart.
Signs, symptoms and risks
Some people with hypertrophic cardiomyopathy don’t have symptoms while others may only feel symptoms with exercise or exertion. Some people may not have signs or symptoms in the early stages of the disease but may develop them over time.
Knowing the signs and symptoms of HCM is important. It can help with getting an early diagnosis when treatment may be most effective.
Signs and symptoms of HCM include:
- Chest pain, especially with physical exertion
- Shortness of breath, especially with physical exertion
- Fatigue
- Arrhythmias (abnormal heart rhythms)
- Dizziness
- Lightheadedness
- Fainting (syncope)
- Swelling in the lower part of your body (ankles, feet, legs) or in neck veins.
HCM is a chronic disease that can get worse over time. This can lead to poorer function and quality of life, long-term complications and more financial and social burden.
As HCM progresses, it can cause other health problems. People with HCM are at higher risk for developing atrial fibrillation, which can lead to blood clots, stroke and other heart-related complications. HCM may also lead to heart failure. It can also lead to sudden cardiac arrest, but this is rare.
HCM has been regarded as the most common cause of sudden cardiac death in young people and competitive athletes in North America, although it is rare.
Getting a diagnosis
Hypertrophic cardiomyopathy is often inherited and is a common form of genetic heart disease. It can happen at any age, but most receive a diagnosis in middle age.
It’s estimated that as many as 1 in every 500 young people in the United States have HCM, but a large percentage of people are undiagnosed. Of those diagnosed, it is estimated that two-thirds have obstructive HCM and one-third have non-obstructive HCM.
A cardiologist or pediatric cardiologist often diagnoses and treats HCM. You may also be referred to a cardiomyopathy center where the health care team has specialized training.
HCM is diagnosed based on your medical history, family history, a physical exam and diagnostic test results.
Print a discussion checklist to make the most out of your appointment. (PDF) | Spanish (PDF)
Fill out a discussion guide online to print and take to your appointment. (PDF) | Spanish (PDF)
Medical and family histories
Knowing your medical history and any signs and symptoms you may have is an important first step. Because HCM can be passed from parents to children, your health care professional will also want to know if anyone in your family has been diagnosed with HCM, heart failure or cardiac arrest. If someone has been diagnosed with HCM, first degree relatives which include siblings and parents, should also be checked. Learn more about genetic testing.
Physical exam
Your health care professional will listen to your heart and lungs with a stethoscope. If they hear a swishing or whooshing sound called a murmur, that could mean there is problem with blood flow through the heart which may suggest HCM.
Diagnostic tests
Diagnosis is typically done by echocardiogram. It checks the function and thickness of the heart muscle and how the blood flows through the heart. In some cases, another type of echocardiogram, transesophageal echo (or TEE), may be performed. A TEE is done using a probe inserted in the throat while the patient is under sedation.
Other diagnostic tests may include:
- Electrocardiogram (ECG)
- Cardiac magnetic resonance imaging (cardiac MRI or CMR)
- Cardiac CT
- Stress tests
- Holter and event monitors to detect abnormal heart rhythms (arrhythmias)
- Genetic testing
HCM Imaging (PDF) | Spanish (PDF)
Diagnostic procedures
Confirming diagnosis or preparing for surgery may also involve one or more medical procedures during cardiac catheterization.
Treatment and management of HCM
There is currently only one disease-specific medication to treat hypertrophic cardiomyopathy. Mavacamten is used to treat the obstructive form of HCM in people who have symptoms.
For people with HCM, a heart healthy lifestyle which includes staying active, eating a healthy diet, maintaining a normal weight, getting good quality sleep and not smoking is recommended. If you have other medical conditions like high blood pressure or diabetes, it’s important that these are managed to avoid heart complications which can develop if left unchecked.
For those with symptoms, the focus is on symptom management using medications and procedures.
Medications
It's important to take the medications that have been prescribed by your health care professional. These medications may help improve symptoms and function, but may have side effects. Medications that may be prescribed for HCM include:
May improve symptoms
May improve symptoms and function
- Myosin inhibitors
Unlike other HCM medications, the cardiac myosin inhibitor mavacamten is used to improve symptoms and function in people with the obstructive type of HCM who have mild to moderate symptoms with activity. Unlike other medications used to treat HCM, mavacamten targets the underlying cause of obstructive HCM.
Procedures
A range of surgical and nonsurgical procedures can be used to treat HCM:
- Septal myectomy – Septal myectomy, also called septal reduction therapy, is open-heart surgery. It’s considered for people with obstructive HCM who, despite taking HCM medications, continue to have severe symptoms. A surgeon removes part of the thickened septum that’s bulging into the left ventricle. This eliminates the obstruction and restores blood flow within the heart and out to the body.
- Alcohol septal ablation (nonsurgical procedure) – Also called nonsurgical septal reduction therapy, alcohol septal ablation is a procedure where ethanol (a type of alcohol) is injected through a tube into the small artery that supplies blood to the area of heart muscle thickened by HCM. The alcohol causes these cells to die. The thickened tissue shrinks to a more normal size. The risks and complications of heart surgery increase with age. For this reason, ablation may be preferred to septal myectomy in older patients with other medical conditions.
- Cardiac implantable electronic devices (CIEDs) – Depending on specific patient risk factors, there are devices that can be implanted in the body to help the heart work better, including:
- Implantable cardioverter defibrillator (ICD) – An ICD helps maintain a normal heartbeat by sending an electric shock to the heart if an irregular heartbeat is detected. This may reduce the risk of sudden cardiac death.
- Pacemaker – This small device uses electrical pulses to prompt the heart to beat at a normal rate. This is used for people with a heart rate that is too slow.
- Cardiac resynchronization therapy (CRT) device – This device coordinates contractions between the heart’s left and right ventricles.
- Heart transplant – In HCM patients with advanced, end-stage disease, a heart transplant may be considered. In this procedure, a person’s diseased heart is replaced with a healthy donor heart.
Prognosis
The long-term outcome for people with HCM is good and most patients with HCM have normal life expectancy without significant limitations or complications.
A small number of people with HCM, however, are at risk for complications including heart failure and sudden death. The risk of heart complications can vary between families and among different members of the same family. It’s important that your health care team identifies people who are most at risk for these complications so preventative treatment can be provided.
Shared-decision making
For people with HCM or at risk for HCM, shared decision-making is recommended in developing a care plan. This includes decisions about genetic evaluation, physical activity, lifestyle and therapy choices.
This should involve thoughtful dialogue among patients, families and their care team discussing the:
- Testing and treatment options
- Risks and benefits of those options to the patient
- Patient’s personal preferences and goals for their treatment plan
- Caregiver’s goals and concerns
Children and HCM
Most children have no symptoms, while others may have mild to severe symptoms including heart failure or sudden cardiac arrest. Children under age 1 more often have symptoms of congestive heart failure. Older children may be symptom-free and unaware they have HCM. Mild symptoms of heart failure can also look like asthma.
Symptoms in infants may be more difficult to detect. They may include:
- Difficulty breathing
- Poor growth
- Excessive sweating
- Crying and agitation during feeding
Many children with HCM can lead relatively normal lives once they are diagnosed and appropriate treatments are started. However, an HCM diagnosis does affect several areas of a child’s life. Specific recommendations should be developed by the child’s health care team.
Support That Lifts You Up







